
Credit: GitHub / Wikipedia. Licenza: Expat License.
Una pandemia dentro la pandemia. Così Ewan Birney, direttore del prestigioso European Molecular Biology Laboratory, ha definito la diffusione della variante B.1.1.7 in una conversazione su Twitter. Dello stesso avviso è Adam Kucharski, epidemiologo alla London School of Hygiene and Tropical Medicine, che in un articolo a sua firma sul Financial Times ha spiegato quali sono i pericoli legati all’emergere di questa variante del SARS-CoV-2 e delle altre che sono state rilevate nelle ultime settimane.
La B.1.1.7 è una variante del virus rilevata per la prima volta il 20 settembre del 2020 nel Kent, regione sudorientale dell’Inghilterra, e diventata a dicembre la variante dominante in tutto il Paese. La variante possiede mutazioni in alcuni aminoacidi della proteina spike, la proteina responsabile per l’ingresso del RNA virale nelle cellule dell’ospite, che la renderebbero più capace di infettare. Il risultato, a livello clinico ed epidemiologico, sembra essere quella di un virus non più virulento (ciò che non causa una malattia più grave) rispetto alle varianti storicamente più diffuse in Europa, ma più trasmissibile. Una stima dell’Imperial College dice che il suo indice di riproduzione potrebbe essere dal 40% al 70% maggiore rispetto a quello della variante storica (tra il 50% e il 74% secondo questo studio della London School of Hygiene and Tropical Medicine). Per quanto riguarda l’efficacia dei vaccini sono stati realizzati diversi test in vitro, sia col siero di pazienti guariti dal COVID-19 che di pazienti vaccinati con il farmaco di Pfizer/BioNTech, che hanno mostrano che le attuali formulazioni dei vaccini hanno efficacia di neutralizzazione rispetto alla B.1.1.7 sostanzialmente invariata. La preoccupazione riguarda però l’aumento dei contagi, delle ospedalizzazioni, che metterebbero nuovamente i sistemi sanitari sotto pressione, e del conseguente numero di vittime.
Per questo motivo il 14 gennaio l’Organizzazione Mondiale della Sanità ha convocato una riunione del COVID-19 Emergency Committee proprio per discutere delle varianti, lanciando un appello agli stati perché incrementino le attività di sequenziamento. Il primo passo per cercare di evitare recrudescenze del contagio è infatti conoscere il grado di diffusione della nuova variante. A fare eco all’OMS è arrivata prima la ECDC, che lunedì ha pubblicato un rapporto che indica le strategie per costruire dei campioni sufficientemente ampi e rappresentativi su cui effettuare le indagini genomiche, e poi la Commissione Europea, che ha chiesto agli Stati membri di sequenziare tra il 5% e il 10% dei campioni positivi.
A che punto sono i Paesi europei rispetto a questo obiettivo? Tra settembre 2020 e il 13 gennaio 2021 la Danimarca, che conduce il maggior numero di indagini genetiche nella UE, ha sequenziato circa il 15% dei casi diagnosticati, segue il Regno Unito con il 5%, Norvegia e Finlandia con il 2%. L’Italia in questo arco di cinque mesi ha sequenziato lo 0,034%, poco meno dello 0,044% della Francia e dello 0,061% della Germania. Danimarca e Regno Unito si distinguono per aver creato già all’inizio della pandemia dei consorzi per l’indagine genomica sul virus. In Danimarca c’è il Danish Covid-19 Genome Consortium che ogni settimana pubblica un aggiornamento sulle attività di sequenziamento e sulla incidenza delle diverse varianti nei nuovi casi diagnosticati. La B.1.1.7 nelle ultime 6 settimane è passata da un’incidenza dello 0,2% a una dell’8,2% (dato aggiornato al 21 gennaio relativo alla seconda settimana di gennaio). Nel Regno Unito già a marzo è stato costituito il consorzio COG-UK grazie alla proposta di Sharon Peacock, microbiologa alla University of Cambridge, con un finanziamento governativo di 20 milioni di sterline.
Le iniziative di sorveglianza genomica britannica e danese sembrano però uniche nel panorama europeo e forse sono da ascrivere a ragioni storiche e di tradizione scientifica. Ma cosa stanno facendo gli altri Paesi dell’Unione? Un esempio di strategia alternativa e meno costosa diretta però solo verso la variante B.1.1.7 è quella attuata dall’agenzia francese per la salute Santé Publique. Nel suo documento settimanale di sintesi epidemiologica del 14 gennaio, Santé Publique ha pubblicato i dati relativi a un’indagine “flash” condotta su tutto il territorio nazionale tra il 7 e l’8 gennaio. Coinvolgendo 89 laboratori l’indagine ha quantificato il numero di campioni che sottoposti ad analisi tramite i kit diagnostici RT-PCR commercializzati dalla società Thermo Fisher Scientific restituivano risultati “discordanti”.
I kit della società Thermo Fisher si concentrano su tre geni del virus, uno di questi è quello che codifica per la produzione della proteina spike del virus (chiamato gene S). Come ben illustrato in questo articolo sul New York Times, La variante B.1.1.7 presenta otto mutazioni sul gene S, di cui sei sono sostituzioni (aminoacidi che sono diversi rispetto a quelli presenti nella stessa posizione della sequenza genomica della variante storica) e due delezioni (aminoacidi mancanti). Una di queste due delezioni (quella di sei nucleotidi codificanti gli aminoacidi in posizione 69-70) fanno sì che il test RT-PCR della Thermo Fisher non trovi il gene S, ma solo gli altri due, dando un risultato “positivo-negativo-positivo”, il cosiddetto risultato “discordante”.
Nell’indagine francese, che ha coinvolto in totale 97 664 campioni prelevati tramite tampone, 7 465 sono risultati positivi. Nella prima fase è stato quantificata la percentuale di casi che hanno restituito risultato discordante: il 3,8% in media sul territorio nazionale (sono disponibili i dati regione per regione) per un totale di 281 casi. In una seconda fase i 281 campioni con risultato discordante saranno sottoposti a sequenziamento per accertare che si trattasse effettivamente di B.1.1.7. I risultati relativi alla seconda fase non sono stati ancora resi noti. Tuttavia, una stima ricavata dai dati di sorveglianza delle varianti condotta dal Centre National de Référence des virus des infections respiratoires dell’Institut Pasteur indica che il 38% dei campioni con risultato discordante sono riconducibili alla variante B.1.1.7 e ha permesso di stabilire in via preliminare che tra l’1% e il 2% dei nuovi casi diagnosticati in Francia fra il 7 e l’8 gennaio sono dovuti alla variante (più precisamente l’1,4%, ovvero 3,8% per 38%).
A partire da questo dato, il gruppo di epidemiologi computazionali dell’Institut national de la santé et de la recherche médicale (INSERM) coordinato dall’italiana Vittoria Colizza ha potuto stimare, per mezzo dei modelli sviluppati finora e validati sui dati dell’epidemia da marzo a oggi, il grado di diffusione che la nuova variante avrà in Francia nelle prossime settimane. Nello scenario migliore, quello che parte da un indice di riproduzione netta R(t) pari a 1,0 e lo aumenta del 50% per mimare la maggiore contagiosità della variante, Colizza e collaboratori prevedono che la B.1.1.7 sarà dominante a metà marzo e si osserverà un nuovo picco nei ricoveri, fino a 35 000 a settimana, due settimane più tardi. Nello scenario peggiore, quello in cui R(t) sale a 1,2 e la variante ha una contagiosità del 70% superiore, già a metà febbraio potrebbero esserci 20 000 ricoveri a settimana.
L’importanza di un’analisi del genere, che verrà aggiornata non appena saranno disponibili i dati del sequenziamento, è che permette di avere un certo anticipo nella pianificazione delle misure di contenimento. In questo senso è una pandemia dentro la pandemia, sappiamo che la curva crescerà e abbiamo la possibilità di evitarlo, introducendo misure di distanziamento più severe. In Germania da martedì è obbligatorio indossare mascherine chirurgiche usa e getta oppure FFP2 sul luogo di lavoro, nei negozi e sui trasporti pubblici, e la Francia potrebbe presto seguire il sue esempio. Un aumento delle ospedalizzazioni significa un aumento nel numero di decessi. Nel Regno Unito, dove grazie al lockdown iniziato il 4 gennaio, per molti troppo tardivamente, la curva epidemica ha cominciato a scendere ma ieri sono morte 1 826 persone (sappiamo bene ormai come ci sia un ritardo tra curva del contagio e curva dei decessi).
In Italia? Lunedì un piccolo comune nella provincia abruzzese di Chieti, Guardiagrele, è finito sulle pagine del Washington Post. A Guardiagrele, infatti, tra il 24 e il 26 dicembre l’Istituto Zooprofilattico Sperimentale di Abruzzo e Molise (IZSAM) aveva rilevato una serie di risultati discordanti che avevano spinto a sequenziare i campioni per scoprire che si trattava di casi di B.1.1.7. Tutti i casi erano residenti nello stesso comune e non avevano alcun collegamento con il Regno Unito né storie recenti di viaggi all’estero. C’è stata una rapida progressione nel numero di casi tra Natale e l’inizio del nuovo anno, tutti contagi avvenuti in ambito familiare. L’IZSAM ha notificato i casi alle ASL e alla regione, come previsto dal protocollo e ha depositato le sequenze ottenute sull’archivio online GISAID, un portale che permette di condividere con i ricercatori di tutto il mondo le sequenze virali, come fa abitualmente e in particolare dall’inizio di questa epidemia. “Non c’è niente di meno sorprendente che osservare delle mutazioni nel SARS-CoV-2” spiega Alessio Lorusso, virologo dell'IZSAM, “ed è importante monitorarne l’evoluzione tramite il sequenziamento, ma non fine a se stesso. È fondamentale, infatti, che quando vengono rilevate sequenze con un numero significativo di mutazioni queste siano poi sottoposte a ulteriori indagini”. È quello che Lorusso ha fatto anche con la B.1.1.7 isolata dai campioni di Guardiagrele, per capire se fosse neutralizzata dal siero di persone guarite, e lo farà presto con il siero dei vaccinati. I risultati sono confortanti, come confermato da altre pubblicazioni apparse nelle ultime settimane. “Le attività di sorveglianza genomica sono fondamentali, e non devono limitarsi solo alla B.1.1.7 che fortunosamente può essere scoperta con i kit che usiamo correntemente. Ad esempio la variante B.1.351, la cosiddetta sudafricana, è positiva al gene S e dunque non è possibile individuarla nello stesso modo”.
Proprio la variante B.1.351, a cui ci gli scienziati si riferiscono anche con la sigla 501Y.V2, sembra destare qualche preoccupazione in più per via di due articoli pubblicati negli ultimi giorni sull’archivio di preprint biorXiv, il primo firmato da un gruppo di ricercatori del National Institute for Communicable Diseases a Johannesburg e il secondo da un team statunitense. I due lavori, che devono ancora essere sottoposti a peer-review, mostrano che la variante è capace di eludere gli anticorpi contenuti nel sangue di pazienti guariti dall’infezione e che l’efficacia di quelli presenti in persone che hanno ricevuto i vaccini di Pfizer/BioNtech e di Moderna viene ridotta, spingendo a pensare che i vaccini contro SARS-CoV-2 dovranno essere aggiornati prima del previsto. “È fondamentale condividere i dati, questo mi sembra scontato ma importante da ricordare in questo momento. Siamo davanti a una pandemia e l’unico modo di venirne fuori e soprattutto di essere più preparati per le prossime volte è quella di avere una risposta il più coordinata possibile”, ha concluso Lorusso.
L’8 gennaio il Ministero della Salute ha emesso una circolare, firmata dal direttore generale della Prevenzione Giovanni Rezza, che ha l’obiettivo di armonizzare le operazioni di sequenziamento svolte dai laboratori di riferimento sul territorio nazionale, indicando una serie di circostanze in cui effettuare il sequenziamento “tempestivamente”. A parte i casi con storia di viaggio nei Paesi in cui è accertata la presenza di nuove varianti virali, la circolare richiede il sequenziamento nel caso di risultati discordanti, negativi cioè al gene S. Ma include anche i casi di reinfezione, di aumento significativo dell’incidenza o di focolai e di infezione in persone già vaccinate. Per quello che riguarda la sorveglianza, la circolare richiede a ciascuna Regione e Provincia Autonoma di inviare ogni mese 10 campioni positivi casuali ma sufficientemente rappresentativi dal punto di vista geografico. Non sembrano previste, almeno per ora, indagini del tipo condotto in Francia. È sufficiente?
Nel documento pubblicato da ECDC è contenuta un’indicazione del numero di campioni che è necessario sequenziare per stimare con diversi gradi di accuratezza la proporzione di casi dovuti a una certa variante.
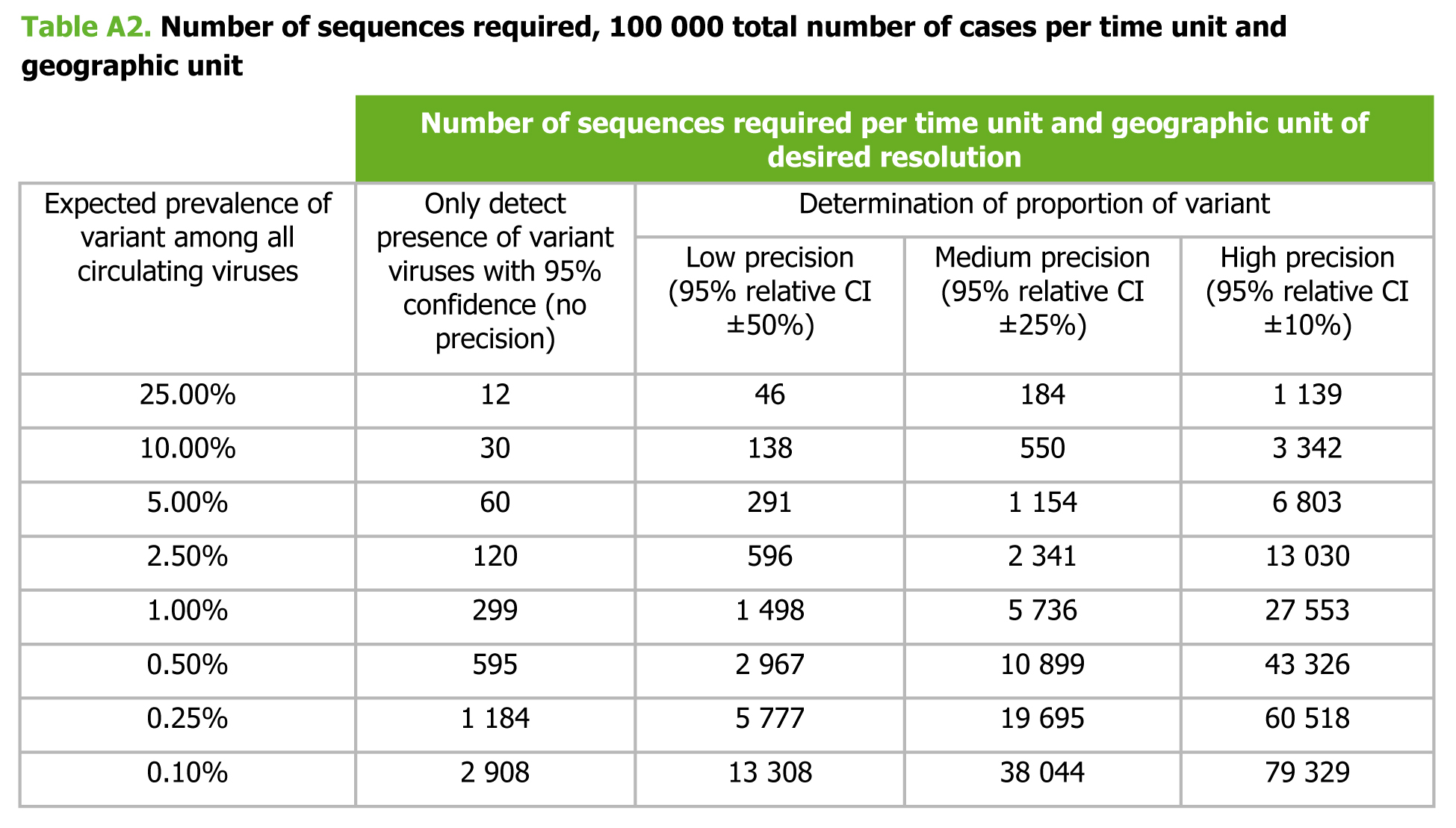
Se un Paese registra 100 000 casi in una settimana e vuole determinare con grado di precisione medio la proporzione con cui una variante con prevalenza attesa dell’1% è presente sul suo territorio dovrebbe sequenziare 5 736 genomi a settimana. Se si accontenta di un grado di precisione bassa, basta sequenziarne 1 498. In Italia, nell’ultima settimana (13-20 gennaio 2021) i casi diagnosticati sono stati 95 130, dunque dovremmo sequenziare dai 1 500 ai 6 000 campioni circa per poter capire quanto circola la B.1.1.7, assumendo che la prevalenza sia simile a quella osservata in Francia. Sarebbero invece sufficienti 300 campioni sequenziati a settimana per accertare che la variante è in circolazione, senza avere alcuna idea dell’accuratezza con cui viene stimate la sua proporzione.
Oltre alla necessità di intensificare le attività di sequenziamento, diversi esperti concordano sul fatto che sia di cruciale importanza accelerare il più possibile la somministrazione dei vaccini. “Dobbiamo fare tutto quello che possiamo ora per vaccinare il maggior numero di persone possibile nel minor tempo possibile, anche se questo significa correre il rischio di selezionare alcune varianti”, ha dichiarato a Science Christian Drosten, direttore dell’istituto di virologia del Charité University Hospital di Berlino, componente del comitato di sette esperti su COVID-19 nominato dalla Commissione Europea e tra gli scopritori del virus responsabile della SARS nel 2003.
Per ricevere questo contenuto in anteprima ogni settimana insieme a sei consigli di lettura iscriviti alla newsletter di Scienza in rete curata da Chiara Sabelli (ecco il link per l'iscrizione). Trovi qui il testo completo di questa settimana. Buona lettura, e buon fine settimana!


{kind=link}